
CHEM-E4106 - Electrochemistry D, Lecture, 9.1.2023-21.2.2023
This course space end date is set to 21.04.2023 Search Courses: CHEM-E4106
Kirja
7. Electrochemical methods
7.2. Steady-state methods
In
steady-state (or stationary) methods, the concentration does not change as a
function of time, i.e.  , and Fick’s second law reduces into the form of
, and Fick’s second law reduces into the form of  . This seems to simplify the solution of the transport
problem, but this does not happen in practice because a true steady-state can only be
reached without convection in spherical geometry. In this case, the
operator
. This seems to simplify the solution of the transport
problem, but this does not happen in practice because a true steady-state can only be
reached without convection in spherical geometry. In this case, the
operator  takes the form
takes the form
 (spherical geometry,
no angular dependency) (spherical geometry,
no angular dependency) |
(7.57) |
|---|
and the concentration profile is
=A+\frac{B}{r}") (spherical
geometry, no angular dependency) (spherical
geometry, no angular dependency) |
(7.58) |
|---|
where A
and B are constants. In linear geometry, in contrast, the
concentration profile would be c(x) = A + Bx, but
the boundary condition c(x→ ∞)
= cb (or 0) would be absurd because the function A + Bx
is not limited. In order to reach a true steady-state, the solution must be
stirred resulting in an unstirred stagnant layer is forming on the surface of the electrode due to adhesion between the electrode and solvent molecules; we denote
the thickness of this layer as  . Now,
the boundary condition changed to c(d)
= cb (or 0), the transport problem has a
meaningful solution in which the concentration changes linearly in the interval
[0,]. The real problem is determining the
thickness .
. Now,
the boundary condition changed to c(d)
= cb (or 0), the transport problem has a
meaningful solution in which the concentration changes linearly in the interval
[0,]. The real problem is determining the
thickness .
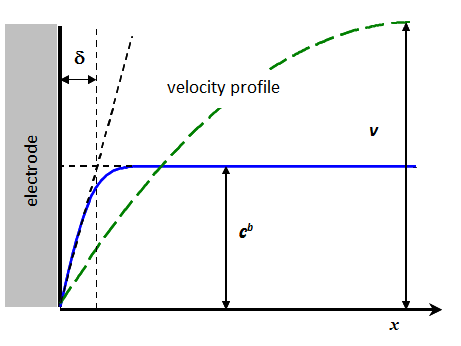
Figure 7.10 shows
how the thickness is usually determined. The
rate of the solution flow changes gradually from zero on the surface (non-slip
condition) to its maximum value in the bulk. In channel flow, for example, the velocity
profile is parabolic. The range of the velocity profile can be an order of
magnitude wider than that of the concentration profile. According to Figure
7.10, is defined as the thickness that is obtained
by extrapolating the linear part of the concentration profile to the distance
where the concentration would be equal to its bulk value. This is, however,
only a thought, a virtual definition, because we cannot measure it. It
is therefore evaluated from the solution of the transport problem using the measurable
limiting current and assuming that in the region 0 < x < there is no convection:
=c^b-\frac{i(\delta-x)}{nFD} \Rightarrow \delta=\frac{nFDc_0}{i_{\text{lim}}}") |
(7.59) |
|---|
In this way, the Tafel slopes and (apparent) rate constant k0 could be determined, as described in Chapter 6, but in practice, steady-state methods are more sophisticated, taking convection explicitly into account via the solution of the convective diffusion equation
 |
(7.60) |
|---|
The solution velocity  must be solved using the Navier-Stokes equation. These methods are therefore also known as hydrodynamic
methods. [1] Because
of the mathematical complexity of flow dynamics, only rotating disk and channel-flow electrodes are introduced in this book.
must be solved using the Navier-Stokes equation. These methods are therefore also known as hydrodynamic
methods. [1] Because
of the mathematical complexity of flow dynamics, only rotating disk and channel-flow electrodes are introduced in this book.
7.2.1 Rotating disc electrode (RDE)
When an electrode is rotated a velocity field is formed in its vicinity in a manner illustrated in Figure 7.11. The Navier-Stokes equation has been solved for this case in the form of a power series; we only need the first parabolic term in the series. The velocity profile perpendicular to the electrode surface is
=-\frac{\omega^3}{v}^{1/2}0.510x^2=-Ax^2") |
(7.61) |
|---|
where  is the angular frequency of rotation (
is the angular frequency of rotation ( ) and
) and  is the kinematic viscosity of the solution (
is the kinematic viscosity of the solution ( , for
water ~0.01 cm2/s at 25 °C). At the electrode, the solution is thrown
away from the center along the surface, but this velocity component does not contribute to the electric current: current is proportional only to the
perpendicular component of the flux. Equation (7.61) also appears to give an
absurd result that the velocity v(x) is increasing without limits with increasing distance x. First, the
full solution with higher order series terms brings the velocity to zero at
long distances and, second, Equation (7.61) applies only in the immediate vicinity of the electrode.
, for
water ~0.01 cm2/s at 25 °C). At the electrode, the solution is thrown
away from the center along the surface, but this velocity component does not contribute to the electric current: current is proportional only to the
perpendicular component of the flux. Equation (7.61) also appears to give an
absurd result that the velocity v(x) is increasing without limits with increasing distance x. First, the
full solution with higher order series terms brings the velocity to zero at
long distances and, second, Equation (7.61) applies only in the immediate vicinity of the electrode.
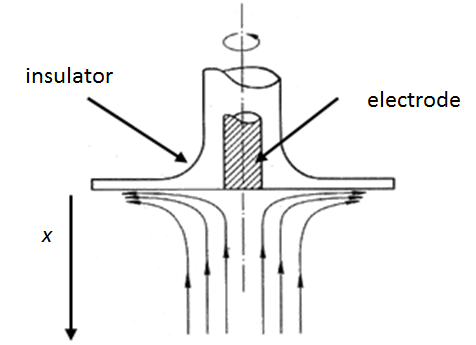
Figure 7.11. Solution flow near a rotating disk electrode.
|
Let us use equation (7.61) as the convection term in the Nernst-Planck equation:
Taking the divergence, the equation of convective diffusion is obtained:
The
boundary conditions are c(
The
integral above can be calculated using the change of variables
where
|
|---|
")
}{D}\frac{dc}{dx}=0")

}{D}p=0")
_0\text{exp}\left(-\frac{A}{3D}x^3\right)")
-c^b= \pm\frac{i}{nFD} \int_{\infty}^{0}{\exp\left(-\frac{A}{3D}x^3\right)dx}")
^{1/3}u^{-2/3}du")
dx}=\left(\frac{D}{9A}\right)^{1/3} \int_{0}^{\infty}{e^{-u}u^{-2/3}du}= \left(\frac{D}{9A}\right)^{1/3}\Gamma(1/3) \approx1.679\left(\frac{D}{9A}\right)^{1/3}")

=m\Gamma(m)=m!")
The limiting current density is obtained from Equation (7.66) setting c(x = 0) = 0:
The limiting current density is obtained from Equation (7.66) setting c(x = 0) = 0:
 |
(7.68) |
|---|
The sign of the
current must be chosen according to the reaction, i.e. if it is cathodic (i < 0) or anodic (i > 0). Equation (7.68) is known as
the Levich equation and it tells us the
important fact that ilim ~1/2. Since all electrode reactions have a limited
rate, an RDE is most suitable for the determination of reaction rate
constants.
If current is measured at a potential where i < ilim, Equation (7.66) can be modified in the form of
)}{\delta}") |
(7.69) |
|---|
where we have
identified the thickness of the diffusion boundary layer as the group of variables
 |
(7.70) |
|---|
Analogously to Chapter 6, we can again write the surface concentration as
=c^b(1-i/i_{\text{lim}})") |
(7.71) |
|---|
Let’s consider an oxidation reaction. Current density is i = nFkox cR(x=0) (only species ‘R’ initially present). Applying Equation (7.71) above, we get the following results:
/i_{\text{lim}}}")  |
(7.72) |
|---|
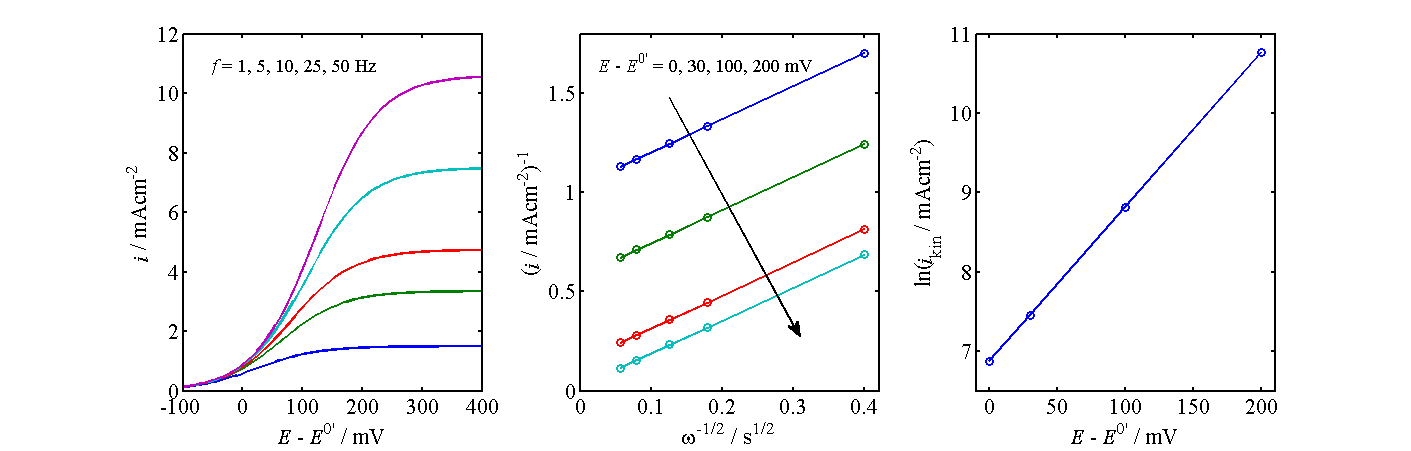
Figure 7.12. Determination of kinetics from the current-voltage curves of an RDE.
The analysis
shown in Figure 7.12 proceeds as follows: At selected potentials, the inverse of
current (density), i-1, is
plotted as a function of  and this line is extrapolated to zero, in other words to infinite angular frequency; the intercept gives the kinetic current
density ikin and thereby kox.
Kinetic current is the current that would occur in the absence of mass
transfer limitations, ikin = nFkox cRb
. A graph representing Equation (7.72) is commonly called as the Koutecký-Levich plot. As shown in the center panel of Figure 7.12, these lines
should have the same slope at each potential. Finally, ln(ikin)
is plotted as a function of the electrode potential. The slope of this line gives
the charge transfer coefficient
and this line is extrapolated to zero, in other words to infinite angular frequency; the intercept gives the kinetic current
density ikin and thereby kox.
Kinetic current is the current that would occur in the absence of mass
transfer limitations, ikin = nFkox cRb
. A graph representing Equation (7.72) is commonly called as the Koutecký-Levich plot. As shown in the center panel of Figure 7.12, these lines
should have the same slope at each potential. Finally, ln(ikin)
is plotted as a function of the electrode potential. The slope of this line gives
the charge transfer coefficient  , and
the intercept at E = E0’ the value of k0.
, and
the intercept at E = E0’ the value of k0.
An RDE has an important modification, a rotating ring-disk electrode (RRDE) in which a concentric ring is placed around the disk at a distance of around 1 mm. Its purpose is to detect species on the ring that are formed on the disk. This can be accomplished, for example, by scanning the disk potential in the cathodic direction and keeping the ring at a constant anodic potential. Any reduced species on the disk are transported immediately to the ring (if they are soluble), where they can be re-oxidized. In this way, reaction mechanisms can be studied when the reaction intermediates are short-lived.
Calculus for the transport problem is rather challenging. An RRDE is calibrated by defining the collector efficiency with a known redox couple, i.e. finding the fraction of the species that is detected on the ring. The collector efficiency depends only on the electrode dimensions and the separation between the disk and the ring. An RRDE requires a bipotentiostat that is capable of controlling the disk and ring potential (or current) separately.
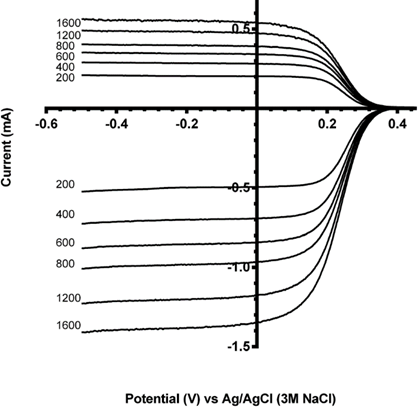
An example of RRDE calibration with a ferro/ferricyanide redox couple is shown below. On the glassy carbon disk, Fe3+ is reduced to Fe2+ whicht is then collected on the platinum ring. The numbers indicate the rotation rate in rpm. The ratio of the ring and disk current, in other words the collector efficiency N ≈ 41%.
7.2.2 Channel flow electrode
Another hydrodynamic method that has a wide practical use is the channel flow electrode. It is often used as a detector in liquid chromatography, capillary electrophoresis or flow injection analysis. The electrode is placed downstream in the flow channel, after a separation unit, and set at a sufficiently anodic or cathodic potential to oxidize or reduce the analyte on the electrode; it is therefore used under limiting current conditions. A reference electrode is usually placed upstream of the working electrode, while a counter electrode is preferably placed downstream of the working electrode in order to prevent its possible reaction products from being transported to the working electrode.
The transport problem of the channel flow electrode can be solved in a closed form for a rectangular channel because the velocity profile is accurately known. In a fully developed Poiseuille flow, the profile is parabolic with zero velocity on the channel walls that is known as the non-slip condition. The limiting current of a rectangular electrode is given by Equation (7.73), also known as the Levich equation:
^{1/3}") |
(7.73) |
|---|
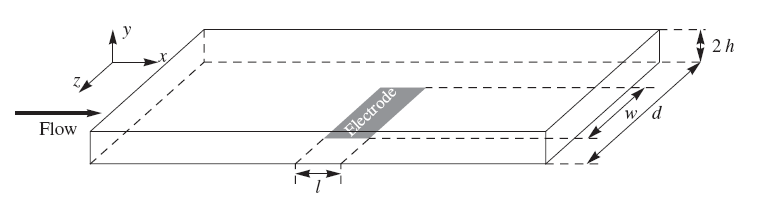
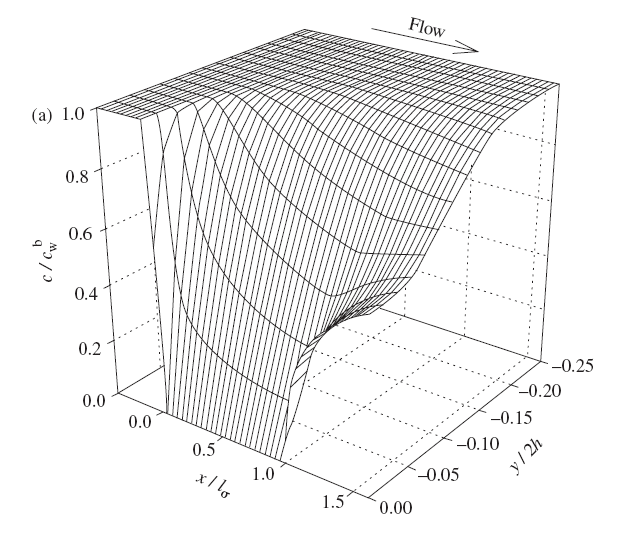
Figure 7.15. Concentration distribution near the channel flow electrode under limiting current conditions. From: Liljeroth et al., J. Electroanal. Chem., 483 (2000) 37.
The coefficient 0.925 in Equation (7.73) is sometimes replaced with 0.835, depending on the simplifying assumptions made in the derivation of the limiting current. The essential feature is, however, that the limiting current is proportional to the cubic root of the volume flow rate. In practice, the electrode response is calibrated using known concentrations, eliminating the significance of the constant coefficients. A detailed description of the solution of the transport problem is available in the literature [3] The problem is to solve first the Navier-Stokes equation and then the convective diffusion Equation (7.60). Figure 7.15 illustrates the concentration distribution of an analyte near the channel flow electrode, solved using the finite element method using Comsol Multiphysics® software. Although all transport problems can today be solved numerically without simplifying assumptions and in an arbitrary geometry, a closed form solution has the advantage that it provides the dependence of, say, the limiting current on the relevant variables explicitly.
[1] A classical textbook of hydrodynamic methods is: V.G. Levich, Physicochemical Hydrodynamics, Prentice Hall, Englewood Cliffs NJ, 1962.
[2] Think: why we are allowed to take the upper boundary condition at x → ∞, instead of x = d?
[3] K. Kontturi, L. Murtomäki, J.A. Manzanares, Ionic Transport Processes, Oxford University Press, 2009.